Prof. Dr. Ulysses Fagundes Neto
Nosso IGASTROPED oferece a realização
do Teste do Hidrogênio no Ar Expirado para o diagnóstico de SIBO com
sobrecarga de Lactulose ou Glicose, para crianças e adultos de quaisquer
idades, bem como para o diagnóstico das intolerâncias aos carboidratos da
dieta, a saber: Lactose, Sacarose, Frutose e Frutanos.
Para agendar a data do exame favor entrar em
contato com a Sra.
Tatianne Rocha no telefone (11) 988427324
Introdução
Vale esclarecer que a sigla SIBO representa
a abreviatura em língua inglesa da entidade clínica Small Intestinal
Bacterial Overgrowth, cuja tradução literal para o nosso
idioma significa Sobrecrescimento Bacteriano no Intestino Delgado.
SIBO é definida “como uma enfermidade na qual o
intestino delgado está anormalmente colonizado por um número aumentado e
anormal de microrganismos da flora colônica”.
A importância do teste
do hidrogênio no ar expirado
O Teste do Hidrogênio no ar expirado
trata-se de um método diagnóstico não invasivo, reconhecidamente de grande
eficácia, desde longa data, pela literatura médica internacional, e cuja
experiência em nossa clínica data desde os primórdios da década de 1980. É
importante enfatizar que este teste não invasivo veio substituir com inegável
vantagem o antigo teste de tolerância, que além de ser invasivo, envolve
inúmeras coletas de sangue de forma sequencial por um longo período.
Mecanismos de proteção
do intestino delgado para evitar a colonização bacteriana
A Figura 1 representa esquematicamente de
forma estilizada a distribuição normal da microbiota do trato digestivo. Em
condições normais ao longo do trato digestivo no sentido craniocaudal ocorre a
presença de bactérias Gram + nas porções altas do intestino delgado, não
superando a concentração de 104 colônias por mililitro de secreção
jejunal. À medida que nos aproximamos do intestino grosso, em particular no
íleo terminal a flora bacteriana aumenta e começam a surgir as bactérias
anaeróbias Gram negativas. Ao cruzar a válvula ileocecal a população
bacteriana colônica aumenta significativamente alcançando cerca de 1012 colônias
por ml, que é praticamente idêntica a encontrada nas fezes.

Figura 1- Modelo esquemático da distribuição normal da microbiota do trato digestivo.
O intestino delgado sadio utiliza vários
mecanismos protetores para evitar a colonização bacteriana pela microbiota
anaeróbica (Figura 2).
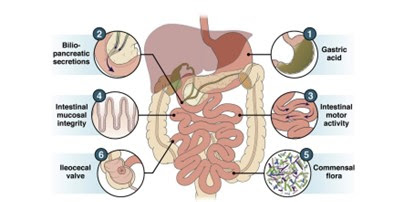
Figura 2- Fatores de proteção contra a ocorrência de SIBO em condições normais e que podem ser suscetíveis de ruptura em situações de enfermidade.
As secreções, ácida gástrica, biliar e
pancreática, inibem a proliferação bacteriana tanto a ingerida quanto aquela
presente na orofaringe que deve migrar distalmente. O peristaltismo do
intestino delgado impede a estase e o crescimento bacteriano. A mucosa
intestinal inclui uma camada de muco e mecanismos antibacterianos intrínsecos,
tais como, as defensinas e imunoglobulinas. Finalmente, a válvula ileocecal
limita o movimento retrógrado das bactérias anaeróbicas colônicas.
Quando e como realizar o
teste para SIBO?
A SIBO é uma síndrome clínica que pode
estar associada a inúmeras enfermidades (fibrose cística, doença de Parkinson,
escleroderma, diabetes e enfermidades do tecido conectivo) e, em transtornos da
interação entre o cérebro e o trato-digestivo, tais como, a síndrome do
intestino irritável (SII), dispepsia funcional, alterações da motilidade
intestinal após cirurgia e uso de opioides e corticosteroides. Os pacientes
portadores de SIBO comumente relatam flatulência e o padrão ouro de
referência para o diagnóstico seria a aspiração da secreção do intestino
delgado com a respectiva cultura bacteriana. Entretanto, levando em
consideração o custo, o caráter invasivo e as dificuldades técnicas para a
obtenção destas amostras, está formalmente indicada a realização do Teste do
Hidrogênio no ar expirado com sobrecarga de lactulose ou glicose para
confirmar o diagnóstico de SIBO.
As principais indicações do Teste do Hidrogênio
no ar expirado estão apontadas na Tabela 1 abaixo.

Teste de sobrecarga com Lactulose
A Lactulose é um dissacarídeo
sintético composto por galactose-frutose, portanto não digerível e não absorvível,
visto que o intestino humano não possui nenhuma dissacaridase capaz de
hidrolisar este carboidrato, e, como consequência a Lactulose é sempre
fermentada pela microbiota intestinal. Neste caso deve-se utilizar a dose
de 20 gramas do carboidrato diluído em solução aquosa a 10%.
Normas para a realização do Teste do Hidrogênio no
ar expirado com Lactulose
O teste deve sempre ser iniciado com a
obtenção da amostra de jejum para a mensuração do H2 no
ar expirado. Vale ressaltar que o paciente deve estar em jejum pelo período de
ao menos 4 horas. Após a mensuração do valor basal de jejum, o qual deve ser,
na imensa maioria dos casos, inferior a 5 partes por milhão (ppm), o teste
respiratório pode começar. O teste com a Lactulose (a
dose é fixa de 20 gramas diluídas a 10% em água) para pesquisa de SIBO deve ser
realizado com a coleta de amostra do ar expirado a cada 15 minutos após a
amostra de jejum durante a primeira hora do teste, e a cada 30 minutos até
completar 2 horas.
Interpretação do Teste do Hidrogênio no ar expirado
A interpretação do resultado do teste
do H2 no
ar expirado baseia-se em 2 fatores fundamentais, a saber: 1- a concentração em ppm
do Hidrogênio no ar expirado e/ou 2- o aparecimento de sintomas após a
realização do teste de sobrecarga.
Caso ocorra a elevação da concentração do H2
acima de 10ppm sobre o nível de jejum, dentro dos 60 minutos iniciais do
teste, indica a presença de SIBO. Essencialmente há 2 possibilidades
para a ocorrência deste perfil particular do H2 no
ar expirado, a saber:
1-
A curva mostra um perfil de 2 picos de
elevação do H2 no
ar expirado, ou seja, 1 deles nos primeiros 30 minutos do teste seguido de uma
diminuição na concentração do H2, o
qual é seguido por nova elevação após os 60 minutos. Este comportamento do
teste indica que há um “Sobrecrescimento Bacteriano no Intestino
Delgado” associado à preservação da válvula ileocecal,
e, também, que as bactérias presentes nas porções altas do intestino delgado
foram capazes de metabolizar a Lactulose. O segundo pico demonstra que a
maior porção da Lactulose foi fermentada no intestino grosso (Figura 3).

2-
A curva mostra um pico
precoce, antes dos 60 minutos após a ingestão da Lactulose, o qual se
mantém por pelo menos em 20 ppm acima do valor basal, sem
apresentar queda no valor do H2 no ar expirado, até os 90 minutos. Esta curva apresenta um
“quase” perfil de 2 picos, sem que ocorra o “vale” entre o primeiro e
segundo picos (Figura 4).

Sobrecrescimento bacteriano no intestino delgado (SIBO) associado a
sintomas ou entidades clínicas que não apresentam evidências de má digestão/má
absorção
Sabe-se que SIBO pode causar diarreia
na ausência de outros aspectos clínicos de má digestão e/ou má absorção
(esteatorreia, desnutrição, deficiências de vitaminas e nutrientes) desde a
metade do século 20, com especial prevalência entre os indivíduos idosos.

Figura 5- Ação patológica da microbiota na SIBO sobre a gordura da dieta.

Figura 6- Ação patológica da microbiota na SIBO sobre os nutrientes da dieta.
Esta hipótese é plausível e se apoia em
vários fatores. Em primeiro lugar, os fatores de risco para SIBO
(hipocloridria, dismotilidade intestinal e resposta imunológica prejudicada) têm
sido descritos neste grupo populacional.

Figura 7- Ação patológica da microbiota sobre a lactase na SIBO.
Em segundo lugar, inúmeros mecanismos podem
ser levados em consideração para explicar a patogênese da diarreia devido à SIBO,
e, a maior parte deles gira ao redor das interações microbiota-sais biliares,
mesmo porque, efeitos diretos do microbioma alterado sobre o sistema
neuromuscular entérico deve ser levado em consideração.

Figura 8- Ação patológica da microbiota sobre os sais biliares na SIBO.
Uma ruptura na fisiologia dos sais biliares
tem sido considerada para explicar casos de diarreia sem causas conhecidas, bem
como, na SII com diarreia. Embora a diarreia relacionada aos sais
biliares tenha sido considerada decorrente de problemas da não reabsorção dos
sais biliares pelo transportador ileal dos sais biliares, interações entre a
microbiota e os sais biliares também têm sido levadas em consideração.
Alterações na composição da microbiota fecal, que provavelmente refletem a
microbiota colônica, têm sido descritas em associação com alterações da
fisiologia dos sais biliares em pacientes com SII-diarreia, porém saber se tais
alterações seriam primárias ou secundárias, necessita a realização de
investigações mais detalhadas.
Uma das maiores alegações controvertidas
relativas à SIBO tem sido sua relação com a Síndrome do Intestino
Irritável (SII). Qual a possível relação entre SIBO e SII,
embora seu nexo seja duvidoso? Tem sido demonstrado que pacientes portadores de
SII, ao realizar o Teste do Hidrogênio no ar expirado após
sobrecarga com Lactulose, estes testes revelaram-se positivos de forma
mais prevalente do que em controles sadios. Além disso, comprovou-se a
ocorrência da normalização dos testes e o desaparecimento dos sintomas após a
realização de um ciclo terapêutico com antibioticoterapia. A eficácia da
Rifaximina como atenuante dos sintomas na SII tem sido considerada uma
evidência que dá suporte para esta associação SIBO e SII.
Alterações nos padrões motores do intestino
delgado poderiam também fornecer alguma base para o conhecimento da
interrelação SIBO e SII. Embora ainda pouco explorada, nos anos
mais recentes, existem evidências que caracterizam uma dismotilidade intestinal
na SII, porém essa hipótese ainda requer confirmação. Presume-se que a
microbiota presente no intestino delgado em pacientes com SII poderia
afetar o sistema neuromuscular entérico, e, por isso, ser a responsável pela
dismotilidade, posto que a SIBO tem sido sugerida ocorrer em um subtipo
de SII, a SII pós-gastroenterite.
SIBO tem ido proposta como responsável por
impactar um outro fenômeno que poderia ser relevante para a SII, isto é,
o envolvimento desde a barreira intestinal juntamente com o sistema imunológico
associado ao intestino, e, pela via do eixo microbiota-intestino-cérebro,
alcançar o sistema nervoso central, e, desta maneira, tornar-se um hospedeiro
de tal cenário, simulando um “transtorno autoimune”.
Referências
Bibliográficas
1-
Bushyhead D & Quigley E –
Gastroenterology 2022;163:593-607
2-
Wurm P et cols. –
JPGN 2023;77:31-38
3-
Moshirea B e cols. – Gastroenterology
2023;165:791-800
4-
Levine A e cols. – Gut 2018;67:1726-38














